
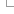
Ensembl Variation - Calculated variant consequences
For each variant that is mapped to the reference genome, we identify all overlapping Ensembl transcripts. We then use a rule-based approach to predict the effects that each allele of the variant may have on each transcript. The set of consequence terms, defined by the Sequence Ontology (SO), that can be currently assigned to each combination of an allele and a transcript is shown in the table below. Note that each allele of each variant may have a different effect in different transcripts.
This approach is applied to all germline variants and somatic mutations stored in the Ensembl databases. The resulting consequence type calls, along with information determined as part of the process, such as the cDNA and CDS coordinates, and the affected codons and amino acids in coding transcripts, are stored in the Ensembl Variation database and displayed on our website. For human and mouse variants any overlap with regulatory features is also displayed. For structural variants consequence terms are calculated on the fly for display on our website or API access. You can use this pipeline to annotate your own data via Ensembl VEP. By default, Ensembl VEP will include upstream and downstream annotations for variants within 5kb of a nearby feature, see --distance in Ensembl VEP options.
See below a diagram showing the location of each display
term relative to the transcript structure:

The terms in the table below are shown in order of severity (more severe to less severe) as estimated by Ensembl, and this ordering is used on the website summary views. This ordering is necessarily subjective and API and Ensembl VEP users can always get the full set of consequences for each allele and make their own severity judgement. The IMPACT rating is a separate rating given for compatibility with other variant annotation tools (e.g. snpEff).
| * | SO term | SO description | SO accession | Display term | IMPACT |
|---|---|---|---|---|---|
| transcript_ablation | A feature ablation whereby the deleted region includes a transcript feature | SO:0001893 | Transcript ablation | HIGH | |
| splice_acceptor_variant | A splice variant that changes the 2 base region at the 3' end of an intron | SO:0001574 | Splice acceptor variant | HIGH | |
| splice_donor_variant | A splice variant that changes the 2 base region at the 5' end of an intron | SO:0001575 | Splice donor variant | HIGH | |
| stop_gained | A sequence variant whereby at least one base of a codon is changed, resulting in a premature stop codon, leading to a shortened transcript | SO:0001587 | Stop gained | HIGH | |
| frameshift_variant | A sequence variant which causes a disruption of the translational reading frame, because the number of nucleotides inserted or deleted is not a multiple of three | SO:0001589 | Frameshift variant | HIGH | |
| stop_lost | A sequence variant where at least one base of the terminator codon (stop) is changed, resulting in an elongated transcript | SO:0001578 | Stop lost | HIGH | |
| start_lost | A codon variant that changes at least one base of the canonical start codon | SO:0002012 | Start lost | HIGH | |
| transcript_amplification | A feature amplification of a region containing a transcript | SO:0001889 | Transcript amplification | HIGH | |
| feature_elongation | A sequence variant that causes the extension of a genomic feature, with regard to the reference sequence | SO:0001907 | Feature elongation | HIGH | |
| feature_truncation | A sequence variant that causes the reduction of a genomic feature, with regard to the reference sequence | SO:0001906 | Feature truncation | HIGH | |
| inframe_insertion | An inframe non synonymous variant that inserts bases into in the coding sequence | SO:0001821 | Inframe insertion | MODERATE | |
| inframe_deletion | An inframe non synonymous variant that deletes bases from the coding sequence | SO:0001822 | Inframe deletion | MODERATE | |
| missense_variant | A sequence variant, that changes one or more bases, resulting in a different amino acid sequence but where the length is preserved | SO:0001583 | Missense variant | MODERATE | |
| protein_altering_variant | A sequence_variant which is predicted to change the protein encoded in the coding sequence | SO:0001818 | Protein altering variant | MODERATE | |
| splice_donor_5th_base_variant | A sequence variant that causes a change at the 5th base pair after the start of the intron in the orientation of the transcript | SO:0001787 | Splice donor 5th base variant | LOW | |
| splice_region_variant | A sequence variant in which a change has occurred within the region of the splice site, either within 1-3 bases of the exon or 3-8 bases of the intron | SO:0001630 | Splice region variant | LOW | |
| splice_donor_region_variant | A sequence variant that falls in the region between the 3rd and 6th base after splice junction (5' end of intron) | SO:0002170 | Splice donor region variant | LOW | |
| splice_polypyrimidine_tract_variant | A sequence variant that falls in the polypyrimidine tract at 3' end of intron between 17 and 3 bases from the end (acceptor -3 to acceptor -17) | SO:0002169 | Splice polypyrimidine tract variant | LOW | |
| incomplete_terminal_codon_variant | A sequence variant where at least one base of the final codon of an incompletely annotated transcript is changed | SO:0001626 | Incomplete terminal codon variant | LOW | |
| start_retained_variant | A sequence variant where at least one base in the start codon is changed, but the start remains | SO:0002019 | Start retained variant | LOW | |
| stop_retained_variant | A sequence variant where at least one base in the terminator codon is changed, but the terminator remains | SO:0001567 | Stop retained variant | LOW | |
| synonymous_variant | A sequence variant where there is no resulting change to the encoded amino acid | SO:0001819 | Synonymous variant | LOW | |
| coding_sequence_variant | A sequence variant that changes the coding sequence | SO:0001580 | Coding sequence variant | MODIFIER | |
| mature_miRNA_variant | A transcript variant located with the sequence of the mature miRNA | SO:0001620 | Mature miRNA variant | MODIFIER | |
| 5_prime_UTR_variant | A UTR variant of the 5' UTR | SO:0001623 | 5 prime UTR variant | MODIFIER | |
| 3_prime_UTR_variant | A UTR variant of the 3' UTR | SO:0001624 | 3 prime UTR variant | MODIFIER | |
| non_coding_transcript_exon_variant | A sequence variant that changes non-coding exon sequence in a non-coding transcript | SO:0001792 | Non coding transcript exon variant | MODIFIER | |
| intron_variant | A transcript variant occurring within an intron | SO:0001627 | Intron variant | MODIFIER | |
| NMD_transcript_variant | A variant in a transcript that is the target of NMD | SO:0001621 | NMD transcript variant | MODIFIER | |
| non_coding_transcript_variant | A transcript variant of a non coding RNA gene | SO:0001619 | Non coding transcript variant | MODIFIER | |
| coding_transcript_variant | A transcript variant of a protein coding gene | SO:0001968 | Coding transcript variant | MODIFIER | |
| upstream_gene_variant | A sequence variant located 5' of a gene | SO:0001631 | Upstream gene variant | MODIFIER | |
| downstream_gene_variant | A sequence variant located 3' of a gene | SO:0001632 | Downstream gene variant | MODIFIER | |
| TFBS_ablation | A feature ablation whereby the deleted region includes a transcription factor binding site | SO:0001895 | TFBS ablation | MODIFIER | |
| TFBS_amplification | A feature amplification of a region containing a transcription factor binding site | SO:0001892 | TFBS amplification | MODIFIER | |
| TF_binding_site_variant | A sequence variant located within a transcription factor binding site | SO:0001782 | TF binding site variant | MODIFIER | |
| regulatory_region_ablation | A feature ablation whereby the deleted region includes a regulatory region | SO:0001894 | Regulatory region ablation | MODIFIER | |
| regulatory_region_amplification | A feature amplification of a region containing a regulatory region | SO:0001891 | Regulatory region amplification | MODIFIER | |
| regulatory_region_variant | A sequence variant located within a regulatory region | SO:0001566 | Regulatory region variant | MODIFIER | |
| intergenic_variant | A sequence variant located in the intergenic region, between genes | SO:0001628 | Intergenic variant | MODIFIER | |
| sequence_variant | A sequence_variant is a non exact copy of a sequence_feature or genome exhibiting one or more sequence_alteration | SO:0001060 | Sequence variant | MODIFIER |
* Corresponding colours for the Ensembl web displays.
Missense variants may have further annotation on their effect on the protein function, using a number of algorithms.